MEXPRESS
visualize DNA methylation, expression and clinical data
What?
MEXPRESS is a data visualization tool designed for the easy visualization of TCGA expression, DNA methylation and clinical data, as well as the relationships between them. You can find all the details in our publication.
Why?
While there are several other excellent tools available, such as cBioPortal, Xena, or TSVdb, none of them allow you to look at DNA methylation data in relation to its genomic location and other data types quite like MEXPRESS does.
How?
Easy! Just enter the name of the gene or miRNA you are interested in, select the cancer type you are interested in, and click the plot button. MEXPRESS will then plot the corresponding expression, DNA methylation, and clinical data. Done!
Browser support
We advise you to use MEXPRESS with a modern browser like Chrome or Firefox. MEXPRESS does not work in Internet Explorer!
| Browser | Version tested | Supported |
|---|---|---|
| Google Chrome | 71 | yes |
| Firefox | 64 | yes |
| Safari | 12 | yes |
| Opera | 57 | yes |
| Edge | 40 | yes |
| Internet explorer | ≤11 | no |
Getting started
This is the MEXPRESS start screen:

On the left side you will notice the text input field where you can enter a gene or miRNA name (HGNC symbols, Ensembl gene IDs and Entrez IDs are recognized) as well as the list of cancer types you can choose from. If you want to be guided through an example analysis, simply click the "step-by-step example" button above the gene input box. Let's say you are interested in the expression and methylation data of the CDO1 gene in bladder cancer. Simply enter CDO1 in the text field and select BLCA (bladder urothelial carcinoma).
All that's left to do after selecting a gene and cancer type is clicking the button and the data you selected will be visualised:

There is a lot to see on the screen, so let's go over the different parts in detail.
The figure
The most important thing on the screen is the figure that shows you the DNA methylation, expression and clinical data for the gene (CDO1) and cancer type (bladder cancer) you selected. This figure consists of several main parts. From top to bottom you'll see the legend, the clinical data, the expression and copy number data, and the DNA methylation data.
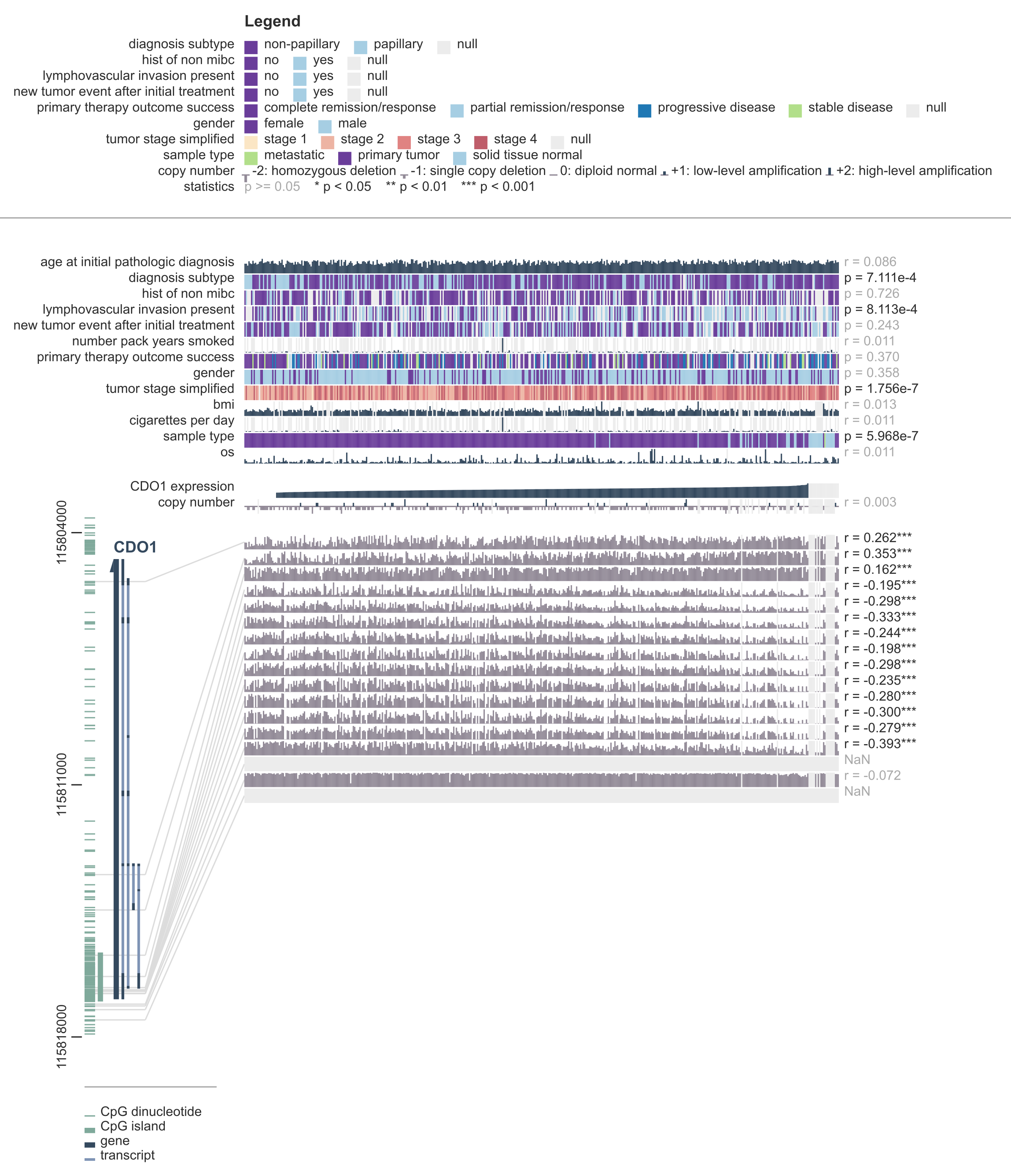
On the left side of the DNA methylation data you can see the genomic annotation. Here, all the genes, transcripts, miRNAs, CpG dinucleotides and CpG islands that are located within the plot window are drawn.
Samples are arranged from left to right. By default, the samples are ordered by the expression of the gene you entered. So in this case, samples with low CDO1 expression are located on the left side of the plot, whereas samples with high CDO1 expression are on the right.
The DNA methylation data are plotted for each probe separately and the data are linked to the genomic location of the probe. If you hover over the DNA methylation data the probe location will be highlighted and if you click on the data, a small box will pop up with some extra information on the probe.
Zooming in
If you are looking at a long gene or a gene with many DNA methylation probes, the plot can become quite large and complex. You can reduce the size of the figure by zooming on a specific part. To do this, you just have to click on the genomic annotation at the left side of the figure and drag a box over the area you would like to zoom in on. You can always zoom out again by clicking the "Zoom out" button.

You can also reduce the complexity of your figure by creating a summary view. We will show you how to do that in the following section.
The toolbar

The toolbar lets you control the plot by filtering and rearranging the samples and by adding or removing data types.
At the top of the toolbar you can see which gene and cancer type you selected as well as the number of samples that are plotted in the figure. Below that, you will find the following options:
- Sort the samples: select a data type from the drop down list to reorder the samples by their value for this data type (remember that by default, the samples are sorted by their expression)
- Filter the samples: select the data type from the drop down list by which you would like to filter the samples. When you select a data type, a new window will pop up with a histogram of the selected data and the different filtering options.
- Select clinical parameters: click the button and select which clinical variables you would like to see in the plot from the full list of all available variables. If you want to go back to the default variables, simply click the "reset" button in the pop up.
- Show the genomic variants: check or uncheck the box to show or hide the genomic variants that are available in the TCGA data. Be aware that for some genes there might not be any variants available.
- Show the summarized view: click this button to generate the summarized view we mentioned in the previous section. In the summarized view, the genomic annotation is drawn horizontally instead of vertically. The samples are split up in different groups based on the variable by which the samples are sorted. If this variable is categorical (for example gender), then the groups will simply match the different categories (male and female). However, if the variable is numerical (for example expression), the samples will be split in two groups based on the median value of the variable (low expression, i.e. expression < median expression, and high expression, i.e expression >= median expression).

- Zoom out: whenever you zoomed in, you can always zoom back out to see the full gene or miRNA by clicking this button.
- Reset the plot: click this button to recreate the default plot.
- Download: select one of the options from the drop down list to download the figure (as a PNG or SVG image), the data (as a text file or in JSON format), or the results of the statistical analysis (as a text file or in JSON format). Note that generating a PNG image might take a few seconds.
- Show information about: select a data type from the drop down list and you will see a pop up window describing this data type in more detail.
Statistics
Another important feature of MEXPRESS is that every figure provides not just a visualization of the data, but also a statistical analysis. The numbers on the right side of the figure show you the p values and Pearson correlation coefficients for the comparison between the variable by which the samples are sorted and all the other variables. Note that all p values you see are Benjamini-Hochberg-adjusted p values, which means that they have been corrected for multiple hypothesis testing.
If we take the default plot as an example, the samples are sorted by their expression, which is a numeric variable. MEXPRESS will then go over all the other variables in the figure and perform the appropriate test. If the other variable is also numeric (for example the DNA methylation data for a specific probe), MEXPRESS will calculate the correlation between the expression data and the DNA methylation data. If the other variable is categorical, MEXPRESS will check the number of categories of this variable. If there are two categories (for example male and female for gender), MEXPRESS will perform a t test check whether there is a statistically significant difference in expression between the male samples and the female samples. When there are more than two categories (for example tumor stage), MEXPRESS will run an ANOVA test to check whether there is a statistically significant difference in expression between the different categories. For the comparison of two categorical variables (for example gender and tumor stage), MEXPRESS uses the chi square test.
We implemented the necessary statistical functions in JavaScript (check the code on GitHub) and we used numerical approximations to calculate the p values, so it is possible that you might obtain slightly different p values when you recalculate them yourself using other tools (such as R).
You can always download the results of the statistical analysis through the download drop-down menu.